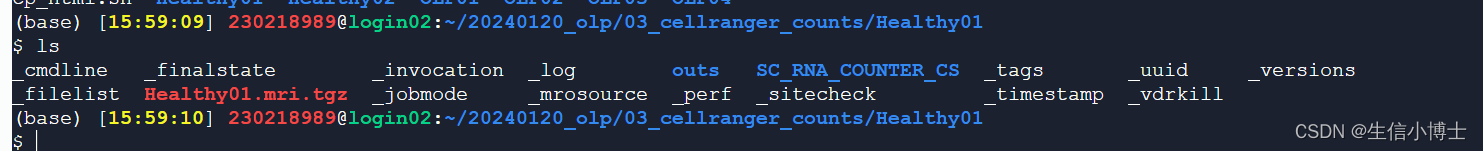
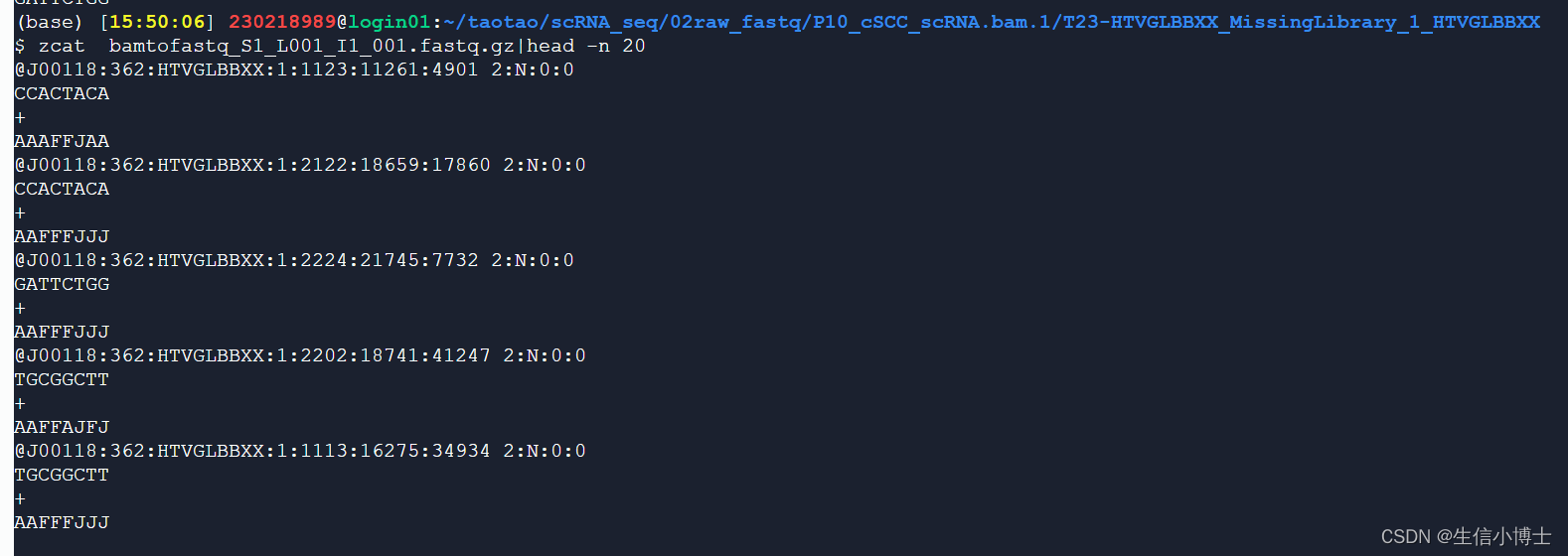
fastq文件
index
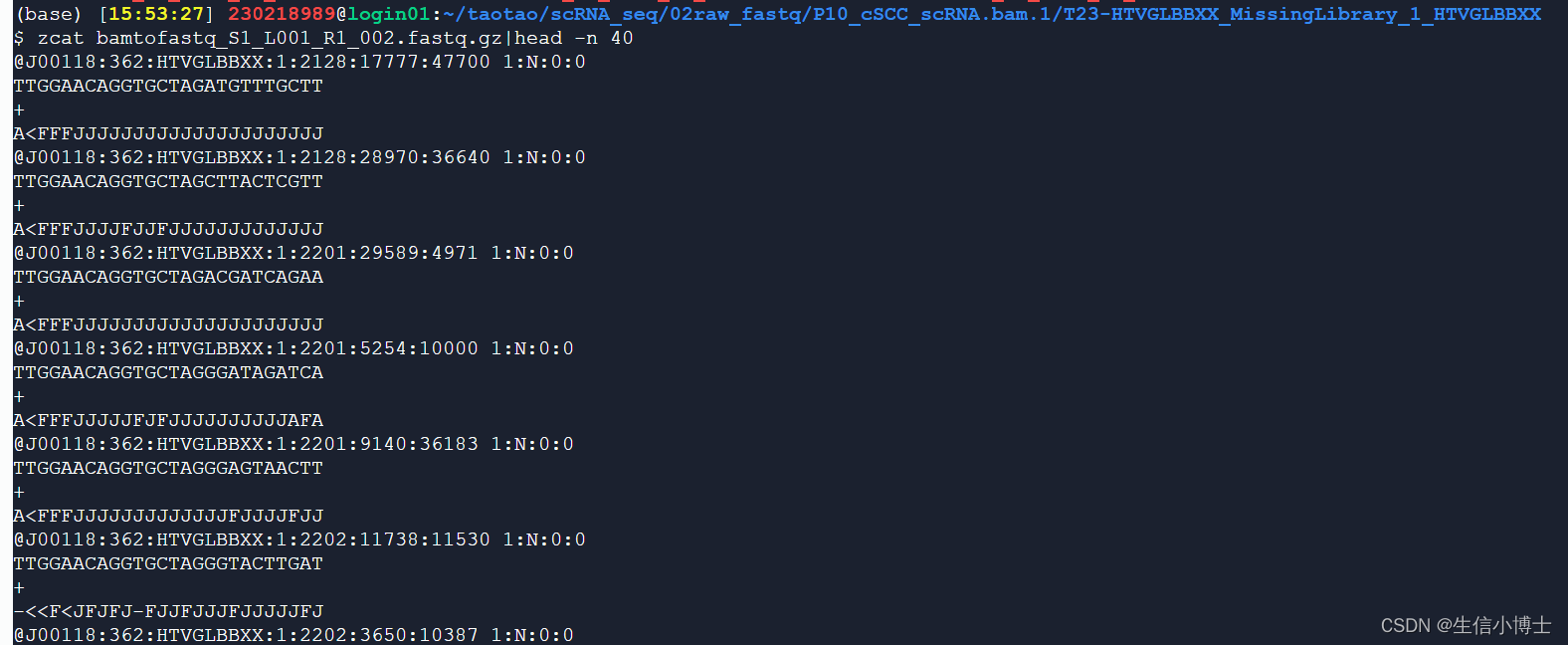
read1
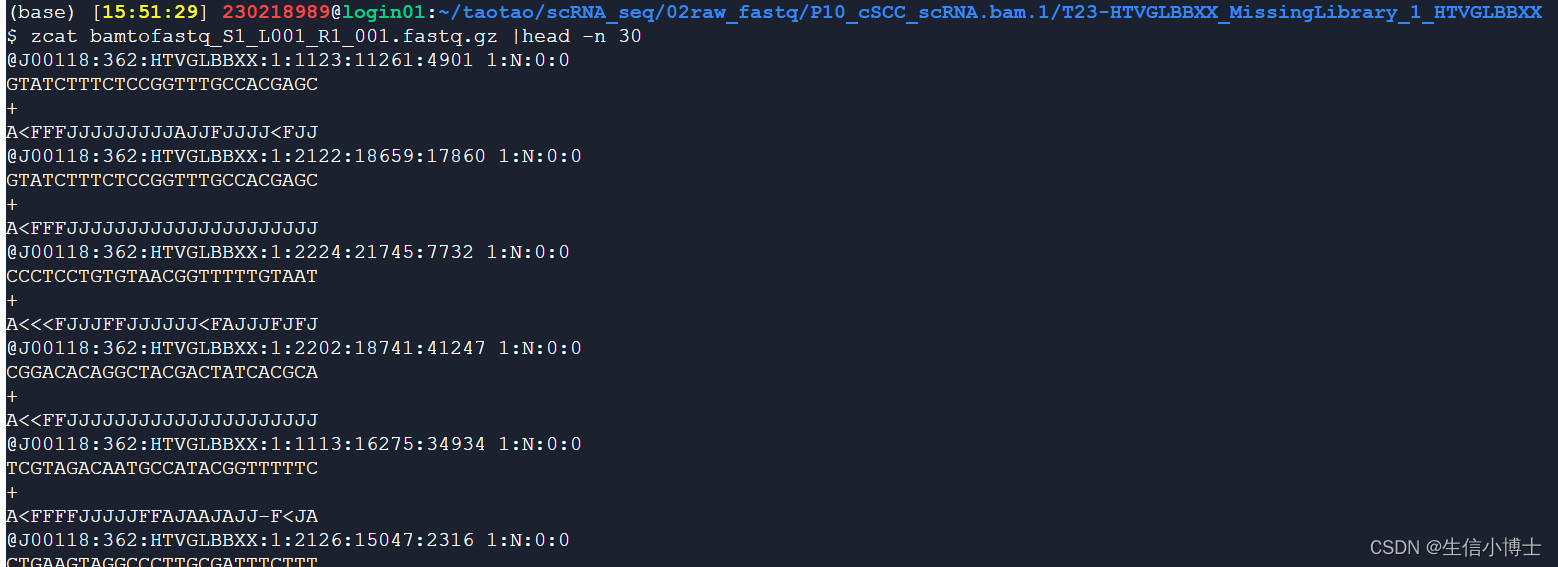
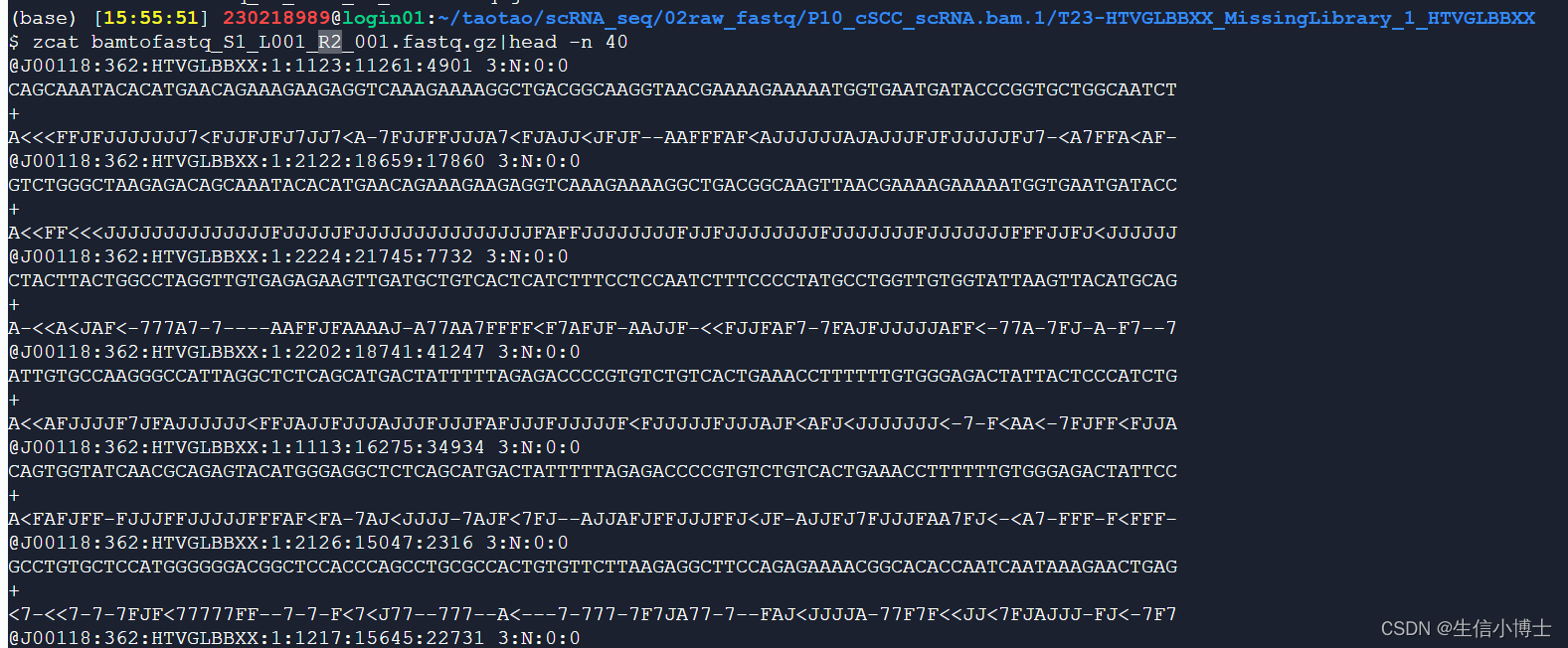
read2
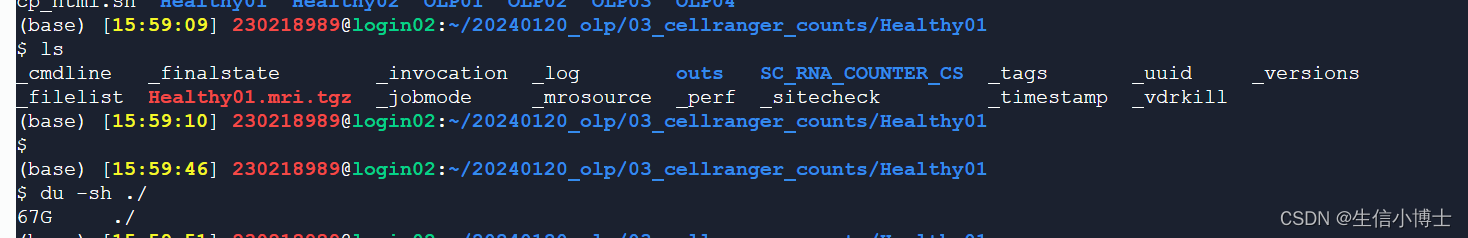
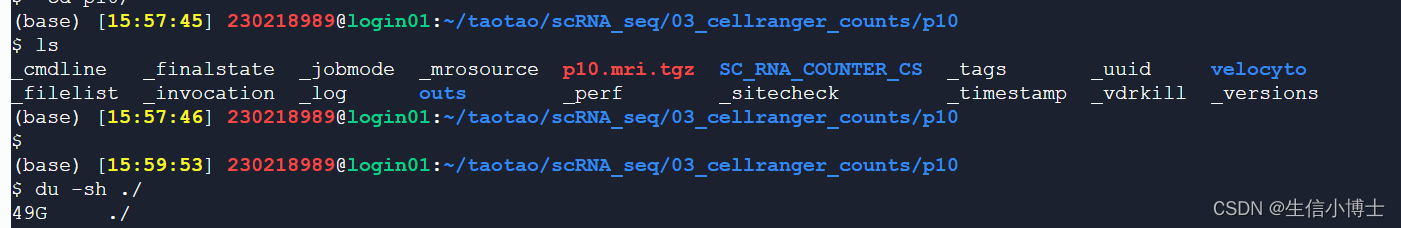
cellraner count 之后
taotao
#p10 ##因为是bam转fastq得到的,且前缀都是bamtofastq
cd /seu_share/home/chaojie/230218989/taotao/scRNA_seq/03_cellranger_counts
ref=/seu_share/home/chaojie/230218989/datasets/cellranger_ref_data/hg19
id=p10
fastqs=/seu_share/home/chaojie/230218989/taotao/scRNA_seq/02raw_fastq/P10_cSCC_scRNA.bam.1/T23-HTVGLBBXX_MissingLibrary_1_HTVGLBBXXcellranger count --id=$id \
--transcriptome=$ref \
--fastqs=$fastqs \
--sample=bamtofastq \
--nosecondary

noredundancy

olp
cd /seu_share/home/chaojie/230218989/20240120_olp/03_cellranger_countsBASE_DIR="/seu_share/home/chaojie/230218989/20240120_olp/01_rawdata" # 修改为你的父目录路径
REF="/seu_share/home/chaojie/230218989/datasets/cellranger_ref_data/human_hg38/refdata-gex-GRCh38-2020-A" # 基因组参考路径# 遍历每个样本文件夹
for SAMPLE_DIR in $BASE_DIR/*; do
if [ -d "$SAMPLE_DIR" ]; then
SAMPLE_NAME=$(basename "$SAMPLE_DIR")
FASTQ_PATHS=""# 遍历样本中的每个子文件夹,并收集路径
for SUB_DIR in "$SAMPLE_DIR"/*; do
if [ -d "$SUB_DIR" ]; then
FASTQ_PATHS="${FASTQ_PATHS}${SUB_DIR},"
fi
done# 移除路径列表末尾的逗号
FASTQ_PATHS=${FASTQ_PATHS%?}# 构建并运行 cellranger count 命令
if [ ! -z "$FASTQ_PATHS" ]; then
echo "Running cellranger count for $SAMPLE_NAME with FASTQs from $FASTQ_PATHS"
echo " "
echo"这是我的运行命令: /seu_share/home/chaojie/230218989/software/cellranger-6.1.2/bin/cellranger count --id=$SAMPLE_NAME \
--transcriptome=$REF \
--fastqs=$FASTQ_PATHS"
echo "\n"
echo " "
echo " "
/seu_share/home/chaojie/230218989/software/cellranger-6.1.2/bin/cellranger count --id=$SAMPLE_NAME \
--transcriptome=$REF \
--fastqs=$FASTQ_PATHS
fi
fi
done